


SMA Research
The Ohio State University is part of a collaborative multi-center effort called Project Cure SMA designed to help find a treatment/therapies for those with SMA. Please contact Sharon Chelnick for more information or to sign up for a trial at columbus@projectcuresma.org. For a complete update on the most current clinical trials at OSU and other participating centers involved in Project Cure SMA visit Project Cure SMA.
Update: September 2022
Using symptomatic Delta7 SMA mice OSU researchers treated them with a combination of an ASO similar to Spinraza ans[sic] another compound
similar to Evrysdi. The 2 treatments worked together to increase SMN and rescue the mice. Spinraza and Evrysdi combined should be
considered for human clinical trial.
Dual SMN inducing therapies can rescue survival and motor unit function in symptomatic Δ7SMA mice.
Update: November 2014
Information about Phase 1 Gene Transfer Clinical Trial for Spinal Muscular Atrophy Type 1 and the Nationwide Children's Hospital Center for Gene Therapy.
Update: February 2012
Dr. Arthur Burghes discusses antisense oligomer treatment in an SMA mouse model in February, 2012. This article contains the transcript as well as a link to the podcast.
Update: January 20, 2012
Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens, the full article, from www.PubMed.gov. The abstract is included below.
Spinal muscular atrophy (SMA) is a leading inherited cause of infant death with a reported incidence of ~1 in 10,000 live births and is second to cystic fibrosis as a common, life-shortening autosomal recessive disorder. The American College of Medical Genetics has recommended population carrier screening for SMA, regardless of race or ethnicity, to facilitate informed reproductive options, although other organizations have cited the need for additional large-scale studies before widespread implementation. We report our data from carrier testing (n = 72,453) and prenatal diagnosis (n = 121) for this condition. Our analysis of large-scale population carrier screening data (n = 68,471) demonstrates the technical feasibility of high throughput testing and provides mutation carrier and allele frequencies at a level of accuracy afforded by large data sets. In our United States pan-ethnic population, the calculated a priori carrier frequency of SMA is 1/54 with a detection rate of 91.2%, and the pan-ethnic disease incidence is calculated to be 1/11,000. Carrier frequency and detection rates provided for six major ethnic groups in the United States range from 1/47 and 94.8% in the Caucasian population to 1/72 and 70.5% in the African American population, respectively. This collective experience can be utilized to facilitate accurate pre- and post-test counseling in the settings of carrier screening and prenatal diagnosis for SMA.
Update: February 4, 2010
According to Dr. Kaspar, Principal Investigator, The Research Institute at Nationwide Children's Hospital: Based on positive pre-clinical studies using a gene therapy to treat a mouse mouse model of SMA, The Kaspar and Burghes Laboratories at Nationwide Children's Hospital and The Ohio State University initiated preliminary studies in non-human primates and have seen successful translation of the gene delivery approach in a larger species. These results have prompted a research and clinical team including Dr. Kissel, Dr. Kolb, and Dr. Mendell to be established to initiate a translational program for human clinical trials for SMA. Presentation of the data and the plans to rapidly move the program forward are forthcoming in the next several weeks. More info coming soon.
Update: August 27, 2009
Breakthrough in the study of central nervous systems: Investigators at The Research Institute at Nationwide Children's Hospital have identified a non-invasive method for delivering genes to the central nervous system, a strategy that penetrates the body's protective blood-brain barrier with unprecedented success. To read more about the research breakthrough visit our research articles page.
Update: October 1, 2008
Greetings SMA families, researchers and friends: We would like to extend our deepest thanks for your dedication and all you have done to help grow support for the SMA Treatment Acceleration Act (H.R. 3334/S. 2042). Thanks to your efforts, 83 Members of the House of Representatives and 21 Senators have signed on to the bill, a major milestone for the first-ever federal legislation authorizing SMA funding.
Our approach and goals for the rest of this year are to continue to push for consideration of the bill. Congress is likely to recess for the fall election season as early as this week, but, there is a possibility that a “lame duck” session will occur following the November elections and before the new Congress and President are sworn into office. If Congress does return for a lame duck session, it is possible that the SMA Treatment Acceleration Act could be considered. As part of an effort to create such an opportunity, our government relations team is in ongoing discussions with Congressional leadership and senior members of the House Energy and Commerce Committee on both sides of the aisle.
Regardless of the outcome of a lame duck session, we have made extraordinary progress over the past 12 months which positions us perfectly to reintroduce the bill at the start of the new Congress next January, continue to grow support, and work towards passage and enactment. Thank you again for your continued hard work as we make progress in securing support for our Act. As we continue our discussions with Congressional leaders in the coming months, we will provide additional updates as well as calls to action to seek your help in moving key decision makers in the House and Senate.
NOTE: If you have any questions about the SMA Treatment Acceleration Act, please feel free to contact any one of our Government Affairs staff: Laura Breiteneicher of the SMA Foundation (laurab@wswdc.com / 202-589-0800), Spencer Perlman of Families of SMA (spencer@fsma.org / 202-333-5750), or Caroline Gibson of Fight SMA (carolinegibson@fightsma.com / 804-515-0080).
Update: August 28, 2008
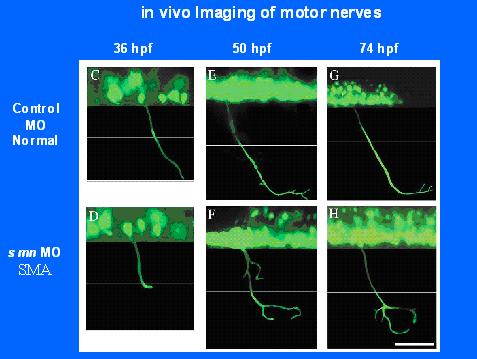
Embryonic motor axon development in the severe SMA mouse: Spinal muscular atrophy (SMA) is caused by reduced levels of survival motor neuron (SMN) protein. Previously, cultured SMA motor neurons showed reduced growth cone size and axonal length. Furthermore, reduction of SMN in zebrafish resulted in truncation followed by branching of motor neuron axons. In this study, motor neurons labeled with green fluorescent protein (GFP) were examined in SMA mice from embryonic day 10.5 to postnatal day 2. SMA motor axons showed no defect in axonal formation or outgrowth at any stage of development. However, a significant increase in synapses lacking motor axon input was detected in embryonic SMA mice. Therefore, one of the earliest detectable morphological defects in the SMA mice is the loss of synapse occupation by motor axons. This indicates that in severe SMA mice there are no defects in motor axon formation however, we find evidence of denervation in embryogenesis.
Read the complete study at Oxford Journals
Vicki L. McGovern1, Tatiana O. Gavrilina1,2, Christine E. Beattie3 and Arthur H.M. Burghes1*
1 Department of Molecular and Cellular Biochemistry 2 Department of Neurology 3 Department of Neuroscience and
Center for Molecular Neurobiology, The Ohio State University, Columbus, OH 43210, USA.
* To whom correspondence should be addressed at: Department of Molecular and Cellular Biochemistry, The Ohio State
University, 363 Hamilton Hall, 1645 Neil Ave, Columbus, OH 43210, USA. Tel: +1 6146884759; Fax: +1 6142924118;
Email: burghes.1@osu.edu
Received May 16, 2008; Accepted July 1, 2008
Update: July 9, 2008
The Ohio State University Medical Center, Department of Neurology is currently enrolling infants with type 1 SMA for the evaluation of Valproic Acid and Carnitine as a potential treatment for SMA. This is a multi center trial fully funded by Families of SMA. For more information on this trial at OSU please contact Shelli Farley at Shelli.Farley@NationwideChildrens.org or call 614-722-2654.
To find other locations for this clinical trial or for more information please visit FSMA. A list of ALL ongoing SMA Clinical Trials can be found at ClinicalTrials.gov.
Update: April 2, 2008
The Kaspar Laboratory at The Research Institute at Nationwide Children's Hospital has an SMA research program that is focusing on therapy development along with basic science studies to understand this disease. The laboratory has specific expertise in gene therapy and stem cell biology. Researchers in the laboratory are currently developing a novel gene therapy that specifically targets motor neurons to replace the gene diminished in SMA. Initial results have demonstrated that we are able to target motor neurons throughout the entire spinal cord efficiently with a one-time dosing, which is the first study to demonstrate this. Current studies are focused on testing this approach in mouse models of SMA. Furthermore, the laboratory has exciting results on defining stem cells, of various types and sources to be directed to differentiate into motor neurons. New studies are being performed to investigate the ability for these cells to regenerate lost motor neurons along with modeling various types of SMA. The laboratory is part of the Center for Gene Therapy and is a translational laboratory that will be able to perform clinical studies using gene therapy vectors by the construction of a new Current Good Manufacturing Practice facility (c-GMP) which is currently in progress. This facility will meet the FDA’s guidelines for production of sterile drug products, allowing our center to produce drug products on-site that are safe enough for human injection.
Update: June 22, 2007
deCODE Chemistry, Inc. and Families of SMA announce the selection of the first ever novel SMA drug compound targeted specifically to treat SMA. Upon gaining FDA approval, the HOPE is to begin clinical trials with this new "Clinical Candidate" within one year. Read the full story
"The SMA Treatment Acceleration Act" will be introduced in the U.S. House of Representatives and the U.S. Senate very soon. The proposed legislation was developed through a collaborative effort among the SMA Foundation, Families of SMA and FightSMA. YOU can help continue their efforts when we announce a "National Call To Action" as soon as the bill is formerly introduced. We will be asking YOU to contact your members of Congress to ask them to join our fight by cosponsoring this bill. More details on the Acceleration Act
Update: April 2007

Carl Reed visits Washington D.C. and makes his way to Capitol Hill visiting with our members of congress and our senator to introduce and encourage their support of the SMA Treatment Acceleration Act and to ask for their continued support of federal funding for SMA. More info on the FightSMA Annual Conference 2007 and SMA Day on Capitol Hill. From left to right: Congressman David Hobson, Carl Reed, Archie Griffin, and Congressman Pat Tiberi |
Update: June 6, 2007
The Ohio State University begins the first clinical drug trial for adults with SMA called THE VALIANT SMA STUDY. For more information and how to enroll, go to Project Cure.
Clinical Trial transcript from the live chat with Dr. John Kissel
2006 SMA Update by Dr. Arthur Burghes
Groups currently in Spinal Muscular Atrophy research at Ohio State and current research areas:
- Arthur Burghes Development of SMA mice, Developing and testing drugs using these mice. Determining when and where high levels of SMN are required to correct SMA and understanding how low levels of SMN cause SMA
- Christine Beattie: Development of SMA fish. Using fish to test drug compounds. Testing which forms of SMN suppress the fish phenotype and what complex is critical for development of SMA.
- John Kissel: Clinical Trials in SMA and characterization of biomarkers for clinical trials.
- Tom Prior: Developed SMA carrier test. Testing for SMA and carrier status. Development of newborn screens. At Columbus Children’s Hospital
- Dawn Chandler: Analysis of SMN2 splicing and factors affecting it, Timing of therapeutic intervention. Reagent sharing
The Fish
The Mice
Testing and drug development in SMA
The moderate SMA mice which live on average to 14 days and were recently published in Human Molecular Genetics and have become the main stay of drug testing in SMA.. We have developed a system that allows oral drug delivery to neonatal mice as early as day 2. We are testing a whole panel of drugs through this system. In particular, we have looked at butyrate and there derivatives that give enhanced stability in animals. These molecules clearly have a beneficial effect in mice by extending survival considerably and enhancing there clinical motor function. In our analysis of this treatment no increase in SMN has been detected raising the possibility that these compounds work independent of SMN induction. We are testing a series of additional compounds in this system and this technique has been transported to other investigators prior to publication and is being used successfully. In addition to these studies it is important to continue more basic studies on drug compounds. How to compounds that induce SMN in culture work? What targets do these drugs act on? It is our goal to find compounds that alter the SMA phenotype and determine how they work this knowledge can be used to find the most effective compounds. In addition to the fundamental studies described above Dr Prior has taken the first steps towards developing an assay for detection of SMA in newborns and Dr. Kissel has been active in the SMA clinical trial of VPA. Thus the work is pushing ahead on many fronts at OSU towards effective and safe treatments for SMA. Although not mentioned by name in this article there are many people (Technicians, Graduate students, Postdoctoral researchers and Clinicians) who have or are working in the various OSU laboratories hard to move SMA research. Research together with families will move progress ahead rapidly towards the goal of safe effective treatment for SMA.
Conclusion
We believe our strength is in innovation. We have developed SMA mice and fish. We stand on the verge of truly understanding the critical function of SMN in SMA. We have drugs that do have an effect on mice. We need to optimize these drugs using basic system and test safe effective drugs in human trials. We have shipped mice, many reagents and techniques to laboratories all over the world. We believe by making all of these available to the neuroscience field that it has and will continue to have a major impact on moving SMA towards a cure. None of this work would have been possible without the Miracle For Madison Fund and several other family funds supporting SMA research at OSU. We attempt to use the Miracle For Madison Fund to pioneer work that is highly innovative and therefore risky. This type of research, is not funded by NIH, but when we are successful and show that it works, we can then apply for additional grant funding which is indeed moving SMA forward.
A Cure Within Reach
A cure is within reach, thanks to the many breakthroughs from these and many other researchers around the world. MFM&F believes the best hope to finding a cure for children and adults suffering from SMA, lies in the commitment of funds toward this research.